
Western blot, a method to separate proteins by size and analyse their relative expression levels, is a much maligned technique of molecular cell biology. The website PubPeer is flooded with evidence of manipulated Western blots, where gel lanes were inappropriately spliced, or where bands digitally duplicated or erased. Some even question the technology as such, since it is indeed mostly Western Blots (and other gels, like Northern Blots for RNA or Southern Blots for DNA or RNA PCR-amplification gels) which are flagged for image manipulation.
It is however not the technology to blame for all the rigging done with it, but the simple fact that it is image based. Anyone with a minimum of image analysis skills or a good eye for duplications can spot Western Blot manipulations. You do not need to be an expert in the technology or even a biologist, to find data rigging. This is exactly why a certain aberration in Western Blot integrity is often either overlooked or dismissed as incompetent nitpicking: the absence of proper loading controls. Just like it seems to be occasionally the case in the publications from the lab of Richard Moriggl, director of the Ludwig Boltzmann Institute Cancer Research and professor at the Medical University of Vienna, Austria. The Moriggl lab studies molecular signalling in cancer cells and tissues, hence its focus on the analysis of regulatory protein phosphorylation and how it changes under various conditions. Such analysis must be always supported by proper gel loading controls, which seems not always be the case here. A reader of my site contacted me with some examples of such inappropriate gel presentation in Moriggl papers, some of which he already posted on PubPeer.

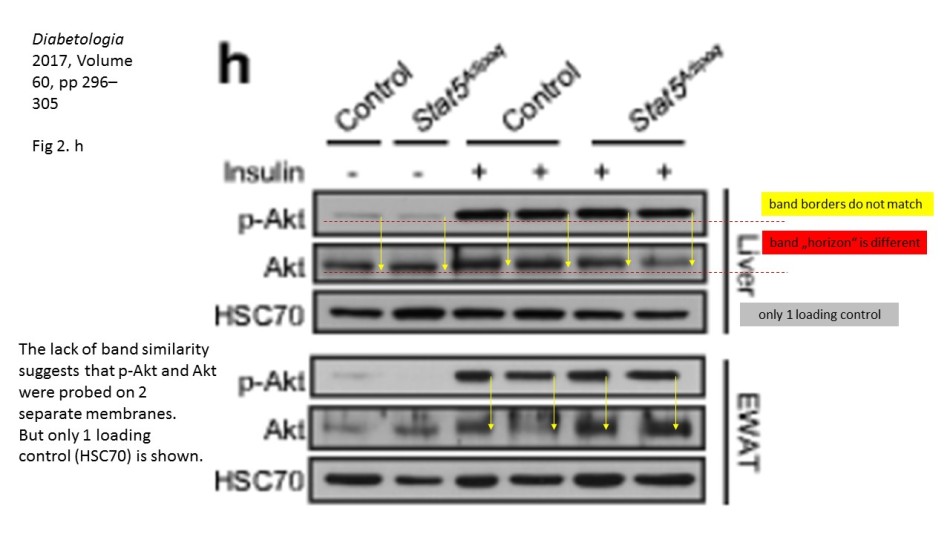
There, a number of Western Blots for a phosphorylated protein seem not to fit the corresponding loading controls provided in the figure. It is not a small issue, because the purpose of such gel analysis is to compare the levels of a certain phosphorylated protein among different samples, say control versus various treatment or a the effect of a treatment at different time points. This is why it is important to ascertain that exactly same amounts of protein-containing cell lysates are present in each gel lane. Here, one does not rely solely on prior protein concentration measurements, but also performs loading controls of the same gel, for a protein which levels are not expected to change in the course of treatments. In the case of phosphorylation analysis, one usually probes with specific antibodies for the same total, non-phosphorylated protein. Only when the equal loading is ascertained can one compare the changing levels of the phosporylation of your protein of interest, or in fact of the overall expression of any other protein you wish to analyse, using corresponding antibodies.
Key point here: it must always be the same gel. It is of no use to detect amazing differences in a protein expression or phosphorylation, while not having a proof that the gel was indeed equally loaded. To probe a different gel where same samples were re-run for an allegedly corresponding loading control does not really make much sense: no two gels are ever exactly same, and a lot of incidental and not so incidental mistakes can happen during the sample loading or the gel run or transfer. This is also why one can spot if the two images really belong to the same physical gel: the lane widths and band sizes and shapes are a give-away if they do not match. Indeed, the sacked cheater Irina Stancheva was caught not only on gel band duplications, but also on using inappropriate loading controls.
Yet the loading controls are often seen as irrelevant. The fallen star of plant sciences Olivier Voinnet built his misconduct defence on his having used a loading control library, where he pulled out a gel image to fit the analysis he aimed to publish. A loading control duplication in a paper from Ronen Alon’s lab at Weizmann Institute was also explained with the use of such libraries (see here). In any case, even if the loading control panel is found to be photoshopped or duplicated, even some established scientists will object: it is just a loading control! The main analytic Western Blot panels are perfectly fine! This is however absolute nonsense, from the scientific point of view. Every Western Blot analysis needs a loading control, done for exactly same gel. Otherwise the entire protein expression analysis becomes utterly useless and one can skip using figures in scientific publications altogether.

Moriggl never replied to my emails. Hence I contacted the Medical University of Vienna with the evidence in his papers, in August 2017 (incidentally, one member of their Scientific Advisory Board is the radiologist and self-plagiarist Hedvig Hricak, from Memorial Sloan Kettering Cancer Center (MSKCC) in New York, read here). The Vice-Rector for Research, Michaela Fritz replied to me the next day:
“Thank you for the information. After consultation with the University of Veterinary Medicine Vienna, the case will be taken up by the Ombudsman of the Vetmeduni, which has already contacted you”.
Indeed, the Ombudsperson of the Vetmeduni Vienna, Mathias Müller wrote to me in parallel:
“Your mail from 23 August 2017 to the management of the Medical University of Vienna was forwarded to the Ombudsoffice of the Vetmeduni. The issues are currently being discussed with the authors. The PubPeer presentations will be addressed with the publishers by submitting point-to-point comments”.
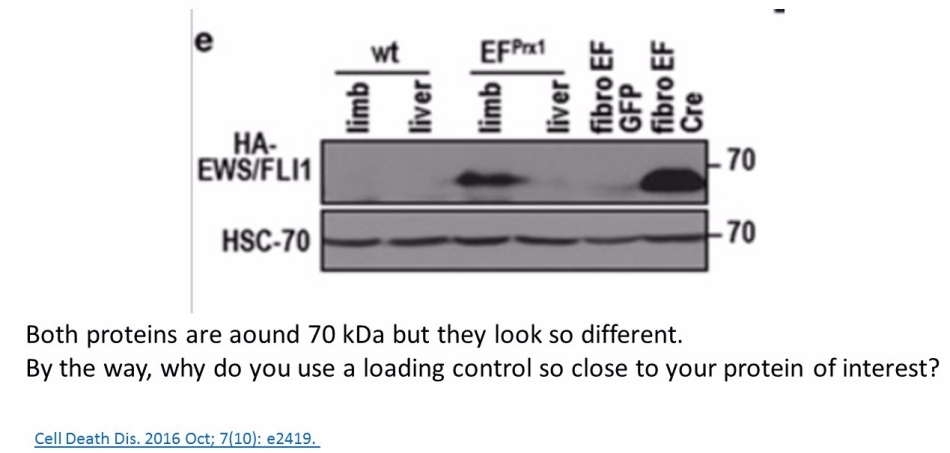
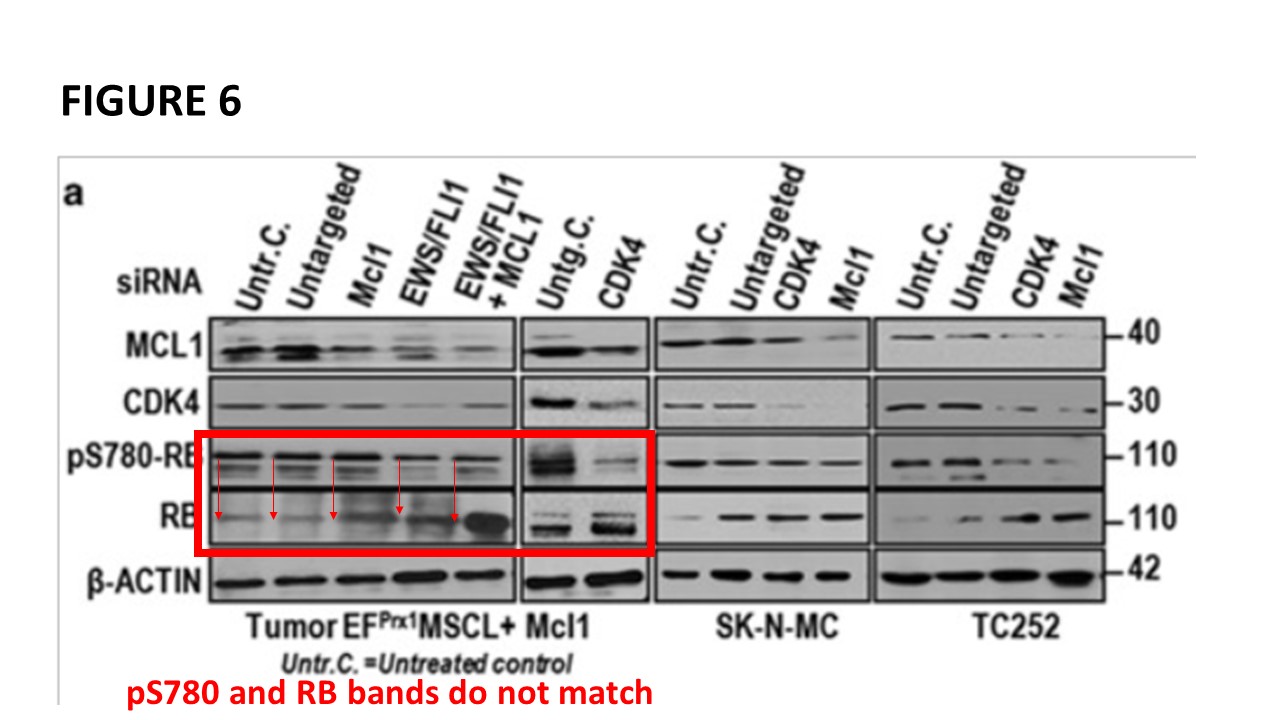

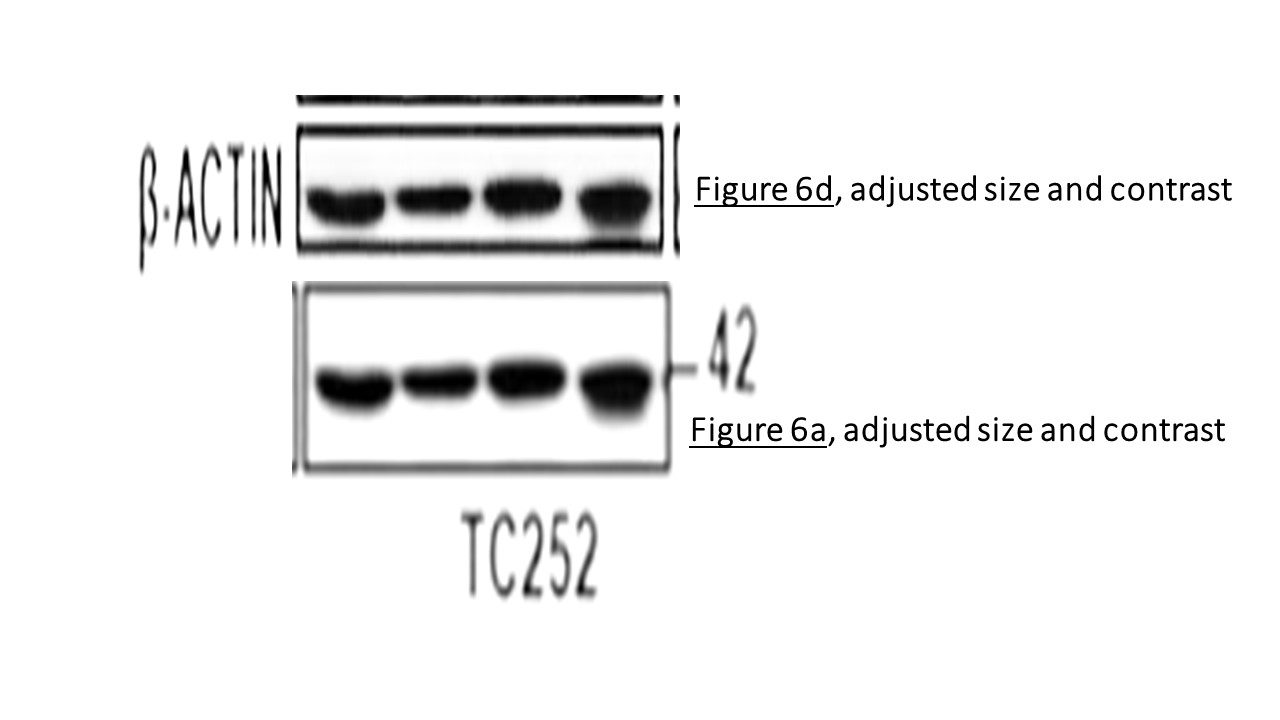
Prior to writing this article, I asked the Ombudsman Müller for an update, but received no reply. There are more inconsistencies in Moriggl papers, like this collaborative paper Gotthardt et al Cancer Discovery 2016 from the neighbouring Vienna lab of Veronica Sexl at the same Medical University Vienna. Interestingly, Sexl has other papers flagged on PubPeer and had already to correct one, namely Kollmann et al, Cancer Cell 2013.

It is of course very likely that correct loading controls will be found and Moriggl will be able to correct his publications to show the matching pairs of gels. Those papers are very fresh after all, the data is certainly readily available. Otherwise readers would be forced to be sceptical about the scientific validity of these Western blot analyses. Without the correct loading control for the exactly same gel, to show the protein loading ratios, a celebrated difference in expression or phosphorylation of a regulatory protein might be nothing but an artefact.
Update 2.11.2017. Matthias Müller, ombudsman of the Vetmeduni Vienna, wrote me this email today:
“Dear Dr Schneider!
With regard to your e-mail dated 23.Aug.2017 to the Rectorate of the Medical University of Vienna, which has been forwarded to the Ombudsman of the Vetmeduni, I can inform you that the issues were clarified with the authors of the publications in Cell Death & Disease and Diabetes.
Replies were made to the PubPeer posts by means of ‘point-to-point’ statements and also sent to the editorial boards of Cell Death & Disease and Diabetes with a request to consider a publication of a corrigendum.
Both Editorial Offices have now approved the Erratum requests and will publish the Corrigenda in the earliest available journal issue”.
Update 13.11.2007. Is the Moriggl lab under investigation by Austrian Science Fund FWF?

Donate!
If you are interested to support my work, you can leave here a small tip of $5. Or several of small tips, just increase the amount as you like (2x=€10; 5x=€25). Your generous patronage of my journalism will be most appreciated!
€5.00


In my opinion the WB is a disgraced old technique which alraedy has good alteranatives such as quantitative mass spectometry and ELISA (which results are not dependent of images). An alternative to cope with the WB technical problems would be the further development of automated WB technology
LikeLike
In ELISA you have no information on the relative size of the protein, where sin WB you do. This can yield a lot of interesting information, as post translational modifications can alter the migration of the protein. Such changes may include phosphorylation (e.g., if you use a phospho site specific antibody, seeing multiple bands indicates that one should look for other phosphorylation sites or modifications, that may occur in tandem.
So it is a great method, but like everything there is a great deal of craft and shortcuts should not be taken.
I would agree that mass spectrometry should be the gold standard, though all to often when submitting a proteomics paper a reviewer asks for some of the discoveries to be ‘validated by WB’. Of course it should be the other way around.
LikeLike
In my opinion:
1. An accurate determination of protein content of the samples and good laboratory practices should allow the researchers to load the same amount of protein in each lane of WB.
2. Work with sample duplicates in the same gel would allow researchers to test their own accuracy in the WB.
3. To load different amounts of protein of the same sample in different adjacent lanes of the gel would allow researchers to test how ‘lineal’ is their effect.
4.A loading control should not change during the experiment. This should be confirmed.
5. When you measure ‘ratios’ of ‘activated’ vs a ‘non activated’ protein you are not measuring ‘activated’ vs ‘total the total protein you are studying. And the ‘total protein’ should be an amount much bigger than the ‘activated protein’ to have a sound measure.
…..
Just some basic rules of basic biochemical experiments that authors, reviewers and editors seem to have forgotten, and could avoid many fake results.
LikeLike
I agree that mass spectrometry should be the gold standard and that after choosing the protein or proteins to study by proteomics we can further analyse these proteins by ELISA (including PTMs if you detect something by mass spectrometry) and also by WB (by following the basic experimental rules)
Nevertheless I think the WB technique can be improved by automating it
Having 3 different techniques at least to confirm the expression of a protein or proteins would be the ideal
LikeLike
Pingback: CNRS hits back at the stream of misconduct evidence – For Better Science
An Erratum in Diabetes for Mueller et al 2017,
Diabetes 2017 Nov; db18er02a. https://doi.org/10.2337/db18-er02a
LikeLike
It means that the blots shown in the paper, actually never existed. New blots were made for this “correction”.
LikeLike
One recent paper from this group has now a correction. Errors with WB images have been admitted, and new WBs have been provided. Diabetes 2017 Nov; db18er02a. https://doi.org/10.2337/db18-er02a
Still the new WBs seem having band shifts, and the loading of the samples is rather different between samples.
LikeLike
Pingback: Collages by Paul Workman, from the Golden Age of Biological Imaging – For Better Science
Pingback: Cardiff investigates two cancer research professors for data manipulation – For Better Science
Pingback: Salk Gandalf Tony Hunter get AACR prize for magic western blots – For Better Science
Cancer Cell. 2005 Jan;7(1):87-99.
Stat5 tetramer formation is associated with leukemogenesis.
Moriggl R1, Sexl V, Kenner L, Duntsch C, Stangl K, Gingras S, Hoffmeyer A, Bauer A, Piekorz R, Wang D, Bunting KD, Wagner EF, Sonneck K, Valent P, Ihle JN, Beug H.
Author information
1
Institute of Molecular Pathology, Dr. Bohr-Gasse 7, A-1030 Vienna, Austria.
Figure 1B. https://imgur.com/0dTiZk8
LikeLike
Pingback: Janine Erler dossiers which ERC does not want – For Better Science
Pingback: How Lopez-Otin et al mocked data policy at Nature Cell Biology – For Better Science
Pingback: Western blot loading control libraries and beyond – For Better Science
Pingback: The Wonderful Adventures of Nils Billestrup with Swedish gels – For Better Science
Pingback: How Andrea Cerutti molested and defiled Journal of Immunology – For Better Science
Pingback: New ERC President Mauro Ferrari was partner of Texas cheater Anil Sood – For Better Science
Pingback: Manchester: “research misconduct concerned only one member of the research group” – For Better Science
Happy to help. Not really my wisdom though, a surmise of the rigorous analysis and conclusions of hundreds of authors across 42 publications, and concluding on what I think is a glaringly obvious solution to this debate going forward – don’t you agree? Those reversible total loading stains are great, really linear. Might even work for the ridiculous GST signals in a pull-down, who knows.
I do hope you will be open minded, objective and not dismissively distract from the point (as above, and I did sign up, you have my email?), stick to the science and refrain from criticizing scientists who, for the many reasons outlined and fully supported above, have most often likely performed more reliable experiments than those that you have been advocating.
LikeLike
Weiss explained nicely to you what you tried at length to muddle, Adrian aged 31 3/4 (you said you don’t mind signing). What I asked you is why you portray me as the evil oppressor and all-powerful persecutor of scientists, and not for example Nature who issue such gel guidelines. Basically, you create a straw man to attack.
LikeLike
42 references, a detailed and I thought clear explanation, and you’re going to use the comment of a person who has clearly not understood or read what I have written (see below) to defend yourself? Classy stuff.
I guess you are just too entrenched down this course to even be at least open minded. I had hoped for much more, a true scientist would not react in this way.
Is the correct way to western blot imposed on us by editors at journals or do they create the guidelines from the debate amongst scientists? It should only be the latter, and your lack of understanding about linear ranges, protein abundance, membrane stripping, consideration of tissue/lineage/confluence, consideration of IPs and pull-downs has led to a campaign for a methodology that the evidence shows is inferior. Why continue to defnd it in the light of all these studies? All the journals have different guidelines, you’re cherry picking the ones you happen to have promoted.
JBC recommends against using house-keeping proteins for instance, and it is time Nature did away with that guideline that every blot needs a houskeeping control – as I’ve described they’re generally meaningless the way you advocate (do you disagree with all of these? PMIDs: 25540176; 24738055; 25852189; 30800670; 25059473; 23709336; 24023619; 18571732; 21186791; 23454168; 24561642 show) and that must be why NRP don’t enforce it at all – in favour of a total protein methodology. Don;t you agree? These do, PMIDs: 18571732; 26004848; 20206115; 23747530; 22929699; 24023619?
All that too much of a muddle for you?
You also seem to want make this personal, revealing my name and age, classy stuff.
LikeLike
It is clearly correct to point out manipulations, duplications, splicings and other irregularities with western blot images on PubPeer and For Better Science. But the unpleasant attacks on scientists for not performing blots in a panel on the same membrane is based on flawed assumptions and non-evidence based thinking. To imply that these scientists are cheating, dishonest or using inferior practices, and calling for corrections or retractions to their articles, is wrong. By all means debate the technical aspects of what is actually a complex technique with many variables so that we can advance and improve western blotting for the future, but please refrain from this uninformed campaign, which is unfortunately beginning to influence publishers, to impose a flawed doctrine based on poorly thought through western blotting strategy. I explain why this thinking is flawed, and suggest a better way forward, below. Thank you for reading.
The call for membrane stripping is unsuitable and less reliable than a parallel gel
Membrane stripping would have been required in the majority of the western blotting experiments criticized on this site in order to achieve the doctrine of “blots from the same gel”. Western blot membrane stripping has been shown to be problematic (PMIDs: 25852189; 19892193; 19523435; 27263489; 11554725; 25059473), and there is no evidence that it is superior to running a fresh, separate gel in parallel when the proteins are a similar size. It is likely inferior, resulting in larger variances when factoring in independent biological repeat experiments. It is difficult to completely remove antibody signals with even harsher stripping buffers, and then it is likely you remove sample protein from the membrane (PMIDs: 25852189; 19892193; 19523435; 27263489; 11554725; 25059473).
Do the accusers propose we all blot for p-AKT T308, strip the membrane, probe for p-AKT S473, strip the membrane, probe for total AKT? What if I want to blot for e.g. 4 or 10 or 30 histone modifications or entire families of 5-10 proteins of similar size on a set of samples? These are very common real world scenarios. Indeed, the more you mess with membranes (strip, reprobe, eat, sleep, wash, re-secondary, wash, re-ECL, develop/scan, repeat), unsurprisingly, the weaker the signal is each time, the patchier the signal, the worse the background and the less reliable they become until you are struggling to see any signal at all (PMID: 19523435).
There are many calls on PubPeer and For Better Science for papers that do not have blots from the same membrane to be corrected or even retracted, which would mean correcting or retracting literally thousands of life sciences papers. Back to the dark ages then. All based on the faith position that membrane stripping (or an alternative) is more reliable than a parallel western blot transfer. I suspect many regard this as nonsense and agree that membrane stripping and alternatives actually raise at least as many concerns and uncertainties than running a gel in parallel, and in fact I can see that they do from the majority of blots in papers. Do you not think we would all be rubbing our hands together with glee at the opportunity to save all those hours and resources if membrane stripping was reliable? There is a reason it is used sparingly and with suspicion, not because we are cowboys and girls trying to cheat you and science with bad controls, but because we really do care about the quality of our western blots and know better.
Given the problems with membrane stripping (PMIDs: 25852189; 19892193; 19523435; 27263489; 11554725; 25059473), the more reliable and diligent approach is surely to run a fresh gel in parallel and probe separately, and then repeat your experiments. We will come on to controlling for loading and transfer in a moment. Other methods may better allow re-use of the same membrane than stripping but these are not widely employed (PMID: 19523435; PMID: 26139277), and recent commercially available stripping buffers claim to be making improvements (of course) but there is little evidence for this as yet.
However, fluorescence based western techniques utilizing differentially labelled secondary antibodies raised against distinct species may allow for more reliable simultaneous (or sequential) blotting, such as phospho and total, and eliminate the need for stripping or a separate gel in some cases (PMID: 24561642). This is becoming more routine. But with the systems available to most scientists you are still limited by the number of species (anyone got 5 µL of dragon anti-acetyl H3 K18 I can pinch?) available, which is typically at best mouse and rabbit for most proteins of interest (POIs). For many experiments you will still not be able to blot for everything on one membrane. Furthermore, as many of you will have experienced, for many antibodies/POIs, fluorescence based methods are just not sensitive enough for detection compared to ECL and film. And has anyone definitively shown that, e.g. for phospho and total, these antibodies do not compete with one another for binding to the same protein on the membrane? Antibodies are huge and other techniques have shown that the ratio of phospho:total is often very low even under stimulated conditions, so you probably would not notice unwanted competition between antibodies.
The call for single loading control blots on every blot is scientifically flawed and risks artifacts
“How do we know about transfer or loading if you don’t do a loading control on each blot?” This has become a persistent, and often vehement stream of somewhat self-righteous criticism, and unfortunately some publishers and guidelines (no doubt in good faith, but also ignorance) have begun to take note. Performing a loading control such as HSP90, GADPH, tubulin or beta-actin on every membrane is not only time-consuming, practically challenging (e.g. size overlap with POIs), expensive and journal space/data-consuming, it is flawed for simple reasons.
Why? Because those proteins are present in your samples at tens, hundreds or thousands of times the level of typical POIs. For instance, across four cell types of different tissue origin the number of copies of HSP90 was ~1000-2000-fold that of AKT1 and ~50 times that of ERK2; copies of GAPDH were ~500-1500 times that of AKT1 and 10-50 times that of ERK2; copies of alpha tubulin (TUBA1B plus TUBA1C) were ~500-800-fold that of AKT1 and 10-25 times that of ERK2 (PMID: 25225357). That really does matter. Many studies have shown that you are very likely out of the linear signal range for these control proteins when you are within the linear signal range for your POI: that is, a large error in loading will appear as little change in signal of your supposed loading control (PMIDs: 25540176; 24738055; 25852189; 30800670; 25059473; 23709336; 24023619; 18571732; 21186791; 23454168; 24561642). How very misleading. For highly expressed proteins, both membrane transfer (proteins binding in layers with only top layer accessible to antibody), and the detection method signal, are vulnerable to saturation, and contribute to the lack of linearity at typical protein loading amounts (23709336; http://tiny.cc/1oq08y). In addition, signals from these proteins are often so intense that they are prone to rapidly “burning out” or quenching, resulting in a halo appearance and other problems, further compounding this lack of utility (PMID: 9509338).
Those who stand next to me in the developing room and do their 0.96 second actin/GAPDH/tubulin exposures, whilst less experienced blotters exclaim “wow I can see the signal with my eyes”, must realise that something is not right. I suspect people know that these “controls” are meaningless but the lanes always came out curiously, and handily, evenly loaded each time, and anyway those guys at PubPeer and For Better Science recommend it so it is all good. No. In order for loading controls like HSP90/GAPDH/beta-actin/tubulin to be useful, in a typical scenario you need to be loading less than 1-10 µg (PMIDs: 24738055; 25852189; 21186791; 23709336; 24023619; 18571732; 30800670). At < 1-10 µg a large proportion of POIs are out of their linear range or undetectable altogether, and therefore separate gels are necessary. But the loading control in this case at least importantly demonstrates that our samples were effectively normalized by the protein assay, instead of being essentially meaningless.
You could spend your time (life) validating novel loading control proteins with a similar expression level/linear range to your protein of interest and probe every blot as appropriate. And again, this saturation and non-linearity problem may be improved versus ECL with fluorescence western blotting techniques, but the problem still very much exists, and these techniques have the alternative downside that high density signals can quench (PMID: 25540176; 25852189; 24561642; 24023619). Trying to control the signal and saturation of signal at the level of antibody concentration is particularly challenging and problematic, and ineffective compared to control at the level of protein loading (PMIDs: 18571732; 26287535; 30054510).
In addition, if you want to compare expression of a POI in different tissues or even different differentiation states within a tissue, using standard loading controls is typically not helpful as the expression of a single control can vary greatly between samples (PMID: 24023619; PMID: 22916200; PMID: 23454168). In fact, even just the confluence of the same cells can disproportionately affect expression of GAPDH and alpha-tubulin, invalidating any comparison or normalisation to the POI (PMID: 20171969); in that scenario you would be better off not normalizing and going with your protein assay.
Now that we have established loading controls as advocated by many on PubPeer and For Better Science are misleading, what does probing for this single protein with outside of the linear signal range tell us about transfer quality? Well, it tells us that some protein was transferred to a narrow sliver of membrane.
Lastly, but really rather importantly, what about IP and pull-down experiments – are these to be dismissed if there are no independent control proteins blotted for on the same membrane? Should I disregard every RAC-GTP pull-down ever performed? Good luck getting GST in to a meaningful range on the same blot. This thinking would again send cell biology back to the dark ages.
But do not despair, as many of you I am sure are aware, there is I think good evidence now for a simple solution to nearly all of these issues.
What is the best way forward?
So if membrane stripping and single standard loading controls cause artifacts, are unhelpful and are inefficient, but there is no chance these guys on PubPeer or For Better Science will trust that our loading and transfers were sound (how do I know these PubPeer types properly covered the membrane with antibody, how do I know their jealous and competitive colleague did not spit in their block, and how do I know they did not accidentally check their mobile phone for a split second in the dark room resulting in perfect artifactual bands, I hear you ask?), what can we do? Well, better results are achieved by monitoring several control proteins at once (PMID: 25852189), but this has obvious issues of practicality. Why not extend this to the entire protein set in your sample? Surely a far superior, rigorous and more versatile method (PMIDs: 18571732; 26004848; 20206115; 23747530; 22929699; 24023619). Well, several companies offer reversible quantifiable total protein stains that you can use to assess and linearly quantify your loading and transfer, either before or after immunoblotting. Alternatively, you can use cheap Coomassie (or alternatively Ponceau) stain, which has been shown to have excellent proportionality with protein level (PMID: 21186791; PMID: 20206115). But I suspect many or most of those criticized did already check their membranes with Coomassie – have we come full circle?
LikeLike
Thank you for teaching your wisdom. Are you so afraid of repressions by “PubPeer and For Better Science” that you are afraid to sign it? If it is other peers and journal editors whose response you are afraid of, why portraying me as the evil all-powerful persecutor of honest scientists?
LikeLike
Do you keep deleting my reply or technical issue?!
Happy to help. Not my wisdom though but a surmise of the rigorous analysis of hundreds of scientists across 42 papers, and then a rather glaringly obvious solution to all of the issues at the end to suggest a way forward. Those reversible total protein stains really are good – have you used them?
I do hope you will be open minded, objective and not distract dismissively from the science as above. Why did you not understand about linear ranges, protein abundance, membrane stripping, consider tissue/lineage/confluence, consider IPs and pull-downs before beginning a campaign for a methodology that the evidence shows is inferior?
LikeLike
you post same comments twice or trice and expect me to keep all versions up? Why?
Just now you replied with an old reply, copy-pasted. Oh the irony.
I am leaving this one up to show how ridiculous your demands to be listened to are.
LikeLike
A normal person would just say “you must be experiencing a technical issue”. Classy
LikeLike
Hi APR, you seem to share the view of Clypeaster Europacificus from PubPeer, https://pubpeer.com/publications/2D084E91EDC85D6C23BC63ACF5E6A8.
I am afraid you do not get the point and your lengthy explanation is not about the concerns raised here. surely you can load your samples on as many gels you wish. However, if you probe AKT in one gel, you need here a GAPDH, actin, or whatever loading control (or if you prefer, Coomassie or Ponceau). And the next blot with p-AKT also needs a loading control. As simple.
Also, as you stated, proteins with similar MW are difficult to detect on the same blot, even after stripping. Why, do you think, authors here https://pubpeer.com/publications/2D084E91EDC85D6C23BC63ACF5E6A8# and here https://pubpeer.com/publications/1C1292425188400AB6A2B0C4B6E2D0 used HSC70 antibody as a loading control, while HSC70 actually has similar MW as AKT and p-AKT? For me it seems that the loading control HSC70 is from a third blot, which has nothing common with the AKT and p-AKT blot. Also, the concerns about two papers from an Austrian professor and a German professor are not only about loading controls. I wonder your opinion about these other concerns.
LikeLike
Hi Weiss.
Unfortunately I feel you did not read or understand my post properly. Please take another look, and look through the ~20 references that have analysed this. GAPDH and other similar controls are expressed at 500-2000-fold more copies per cell than AKT. Therefore, you are very unlikely to have GAPDH and AKT both in their linear ranges when you western blot them on the same membrane; GAPDH signal saturates quickly with total cellular protein over 1-10 ug. The problem is likely even more severe for p-AKT normalised to GAPDH or equivalents because this could be 1/10000-1/1000000 the number of copies of GAPDH. And that’s for a relatively abundant signalling protein that is typically easy to detect (PMID: 25225357). This means referencing or normalizing to GAPDH is meaningless; that is, a large error in loading will appear as little change in signal of your supposed loading control. This has been shown repeatedly, PMIDs: 25540176; 24738055; 25852189; 30800670; 25059473; 23709336; 24023619; 18571732; 21186791; 23454168; 24561642.
At < 1-10 µg a large proportion of many proteins of interest are out of their linear range or undetectable altogether, and therefore separate gels are necessary if you want to blot for GAPDH and e.g. p-AKT. But at least at 1-10 µg the loading control importantly demonstrates that our samples were effectively normalized by the protein assay, instead of being essentially meaningless.
Indeed, I am suggesting to use REVERT or cooomassie total protein stains as a way of drawing a line and going forward with the most rigorous technique. This is the only viable way I can see to confirm transfer and loading on every gel. How does a single loading control band confirm transfer anyway? It’s just a tiny sliver, there could be problems everywhere else.
My main point is you and other should not be going after the scientists who have not done controls on each membrane because you are advocating the inferior method of normalizing to a single houskeeping protein that will inevitably often have saturated signal, and is therefore meaningless.
Where are your references and side-by-side comparisons that show that membrane stripping is superior to running a fresh separate gel in parallel? Membrane stripping or reprobing may be fine in some contexts, yes, but it has been shown to be inefficient and result in loss of bound sample protein, and I would suggest is an inferior method to running a separate gel in parallel (25852189; 19892193; 19523435; 27263489; 11554725; 25059473). This is explained at length above and in the papers, pleas read. So again, I do not think you are in a a position to criticize scientists who run e.g. a separate fresh parallel AKT, p-AKT T308 and p-AKT S473 blots, because this is likely a superior method.
I hope that makes sense.
I am merely debating about western blot theory and technique relevant to the article above and the general technical campaign by Leonid and PubPeer, not fraud or manipulations, which you are of course all doing good work uncovering.
Thanks
LikeLike
Hi APR, I understand your comment and view. However, you oversee my questions and the problem with the Diabetes and Cell Death Disease papers. Maybe we can understand each other better, if you show in these two concrete examples where do you think the proper loading controls are presented.
LikeLike
hi there, you are Jan Peter, right?
LikeLike